
從胺基酸序列預測蛋白質結構 一場持續50年的挑戰
編譯/賴佳昀
序列決定結構
蛋白質是由一串胺基酸折疊而成。折疊的方法複雜曲折、纏繞糾結,但也正是這些三級結構決定了蛋白質在生物體內的功能。誰能釐清蛋白質的結構,誰就能理解生命的基本機制。
以一個貼近我們現在生活的例子來說,開發COVID-19疫苗時,需要瞭解病毒上的刺突蛋白,因為冠狀病毒正是仰賴這種蛋白感染人體細胞。除了新藥的開發,如果把格局再放大一些,蛋白質結構的預測也有助於人工設計出可以分解塑膠垃圾或生產生物燃料的酶,或是用人造蛋白來提高農作物的產量和營養價值。
然而蛋白質的結構分析是非常困難的,傳統上會用到低溫電子顯微鏡(cryo-electron microsopy,cryo-EM)、核磁共振或X光晶體學(x-ray crystallography)。但這些技術非常昂貴,且花的時間久。研究單一種蛋白質便可能要花費數十萬美元的經費和數年時間的反覆實驗,而且不一定適用於所有的蛋白質。已知在各類生物體內扮演不同生化角色的2億種蛋白質中,我們只破解了其中約17萬種的蛋白質結構。
然而從1960年代開始,生化學家Christian Anfinsen證實了胺基酸序列(一級結構)會影響蛋白質的折疊(二級以上結構),並因此獲得1972年的諾貝爾生醫獎。所以理論上可以透過胺基酸的排列順序推出蛋白質的整體結構;但現實其實很骨感,因為影響整體蛋白質折疊的正是這些胺基酸彼此之間的交互作用,若要把所有的可能都條列出來,將會是個天文數字。電腦便是在這個時候被帶了進來。
以AI預測蛋白質結構
1994年,科學家們發起了每兩年一次的「蛋白質預測關鍵測試」(Critical Assessment of protein Structure Prediction,CASP),以競賽的方式促進相關研究的發展。每次比賽,CASP會公布100個左右的蛋白質胺基酸序列,這些蛋白質的立體結構已事先在實驗室中被分析出來,但結果保密。參賽者要在不知情的情況下,單靠CASP所公布的一級結構與自己開發的軟體來預測這100個蛋白質的立體結構。CASP所採用的評分標準為「總體距離測試」(Global Distance Test,GDT),也就是比較預測結果與實際蛋白質結構間的差距,從0(完全不符)到100(完全符合),只要拿到90分以上,便可歸咎於實驗誤差,而不是軟體的問題。
但是一直到2016年,對於較大、結構較複雜的蛋白質,CASP的參賽者成績多在40分左右徘迴,還是參考與競賽題目相似但結構已知的蛋白質才得到的成績。2008年,DeepMind所開發出的AlphaFold首次參戰,也是以同樣的方法獲得了(最困難的題目)60分的佳績,並開啟了AI預測蛋白質結構的風潮;到了2020年,有過半的參賽者都使用深度學習來預測蛋白質結構,使得總體準確度也有所提升。
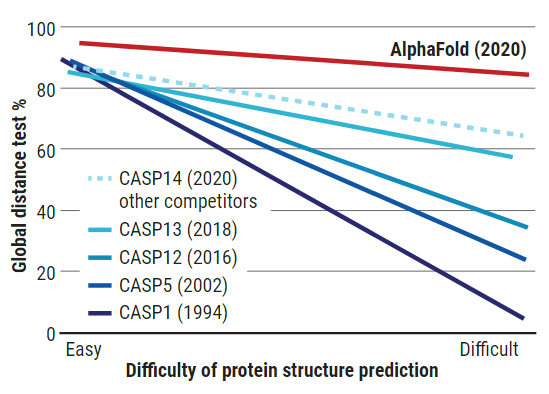
無論是結構相對簡單或複雜的蛋白質,AlphaFold 2020年的預測完勝CASP開辦以來的所有參賽者。(圖片來源:C. BICKEL/SCIENCE)
人紅是非多的AlphaFold
但這樣的成績還遠遠達不到實際應用的水準,AlphaFold的開發團隊於是開始專注於一種稱為「注意力網路」(attention network)的演算法:以公開資料庫中的17萬筆蛋白質資料來訓練模型,比較資料庫中多個序列,並找出在蛋白質折疊過程中常會靠在一起的胺基酸對,然後根據這些結果來推測未知結構中的胺基酸對,同時還能預估每一種猜測的準確度有多高。這就像是在拼拼圖,先拼成一塊塊小片段,再嘗試將每一塊合起來變成一整幅大拼圖。
2020年,AlphaFold在全部100道題目中,達到了中位數92.4的高分;對於最困難的蛋白質,也有中位數87分的亮眼成績(比第二名足足高了25分),其中不乏過去難以X光晶體學破解的蛋白質結構。
這樣的好成績,讓人不免開始懷疑AlphaFold是否在比賽過程中動了些手腳。於是一位評審委員Andrei Lupas特別以自己花了10年心血都還無法破解的蛋白質,專門給AlphaFold另外加考了一道題。那是在一種古細菌上發現的膜蛋白。
AlphaFold最終給出了一個有著三個次單元、中間兩條長長的螺旋臂的蛋白質結構。而這幅AlphaFold所預測的立體結構正好巧妙串起Lupas十年以來的所有實驗數據,讓Lupas心服口服。
魚與熊掌不可兼得
儘管AlphaFold的表現相當出色,但這是以速度作為代價的。另一個使用遞迴幾何網路(Recurrent Geometrical Network,RGN)的演算法,雖然準確率較低,但只要幾秒就能給出結果。對於講求速度的應用,例如細菌或酶的研究,這個算法就會比較合適。
CASP的參賽條件之一,就是所有參賽者必須同意公開演算法的必要細節,使得其他人也可以輕易重製模型,AlphaFold也不例外。
許多的藥物開發都是先透過3D模擬,找出那些能夠插入標靶蛋白活性區位的分子。當然,這也就意味著我們必須先知道標靶蛋白的結構,才能開發出新的藥物來治療例如利什曼病、昏睡病、瘧疾等各種疾病。以人體來說,只有四分之一的蛋白質結構是已知的,剩下的還需要進一步研究。相信隨著模型核心的公開,AlphaFold勢必將在製藥領域掀起另一波革命。
編譯來源
- W. Heaven, “DeepMind’s protein-folding AI has solved a 50-year-old grand challenge of biology“, MIT Technology Review, 2020.
- R. Service, “‘The game has changed.’ AI triumphs at solving protein structures“, Science, 2020.
參考資料
- A. Regalado, “Pfizer’s covid-19 vaccine is highly effective, but don’t expect to get it soon“, MIT Technology Review, 2020.
- A. Senior, J. Jumper, D. Hassabis and P. Kohli, “AlphaFold: Using AI for scientific discovery“, Deepmind, 2020.
(本文由教育部補助「AI報報─AI科普推廣計畫」執行團隊編譯)