晶格能
晶格能 (Lattice enthalpy)
國立臺灣師範大學化學系 趙崇瀚
陰離子與陽離子之間存在庫倫作用力彼此吸引,由於此種異性電荷之間的作用力非常強,比一般的凡德瓦力大得很多,故離子化合物大多是以離子晶體的形式存在。因此當陰陽離子結合形成特定晶體時,其焓值改變量決定了晶體的穩定性
,故可以定義晶格能(\(\Delta H_L\))為:一特定離子晶體分解成其組成的氣態陰陽離子的焓值改變量(式一)。在此定義下,晶格能之值恆為正值,即代表破壞晶格所需的能量大小。
弗洛斯特圖 (Frost diagram)
國立臺灣師範大學化學系三年級 俞姿宇
弗洛斯特圖 (Frost diagram) 或稱自由能 ─ 氧化數圖 (free energy-oxidation state diagram) 是單一種元素的氧化態與自由能的關係圖,單一種元素以不同氧化態存在的分子,在互相比較之下,氧化還原對 (redox couple) 的還原電位,電位差越大代表反應趨勢越強,此種圖示法是很好的判讀工具。
縱軸為電位 \(NE^\Theta\),橫軸為氧化數 \((N)\),氧化數值以遞增或遞減排列都是可以被接受的,值得注意的是橫軸的座標必須合乎線性比例,即使該元素無法存在於所有可能得氧化態。
液相層析(Liquid Chromatography)
國立臺灣師範大學化學系碩士班二年級 翁于婷
層析法是常見且普遍,可將複雜的混合物分離純化的方法。以層析系統的動相分類,主要有氣相層析與液相層析等等1,而液相層析法適用於純化大部分的化合物。
液相層析利用動相 (mobile phase) 和靜相 (stationary phase) 的交互作用,當混合物進入系統內時,不同成分的特性將決定物質在層析管柱內流動的快慢,再利用各成分移動速率之不同來分離2。液相層析儀最主要就是「動相式液體」,所以在液相層析法裡又有:液固層析法 (LSC) 和液液層析法 (LLC)。
液固層析法 (LSC) 或名為吸附層析法,是靜相為細粒固體之表面,溶質與沖提劑競爭固體表面的吸附面積,最適合拿來分離低極性的有機物,也就是最常被使用的高效率液相層析 (HPLC, High-performance liquid chromatography) 的原理3。在這兩相中,通常極性較大的液體會固定吸附在靜相的惰性載體上,而極性較小的液體溶劑則會被當作移動相、極性小的物質先流出,此時稱為「正相層析」(normal phase adsorption)。反之靜相吸附的液體極性小甚至是非極性液體,而移動相為極性液體、極性大的物質先流出,則稱為「逆向層析」(reverse phase adsorption)。
晶場理論 (crystal field theory, CFT)
國立臺灣師範大學化學系三年級 俞姿宇
晶場理論於 1929 年由漢斯·貝特 (H. Bethe) 和約翰·凡扶累克 (J.H. van Vleck) 首先提出。主要用 d 軌域開裂 (splitting) 的情況來解釋錯合物的顏色、磁性、立體構型、熱力穩定性和錯合物畸變。1
錯合物的中心金屬經常是過渡元素,過渡元素具有五種相等能量的 d 軌域,在空間中的方向都不同。首要假設是將配位鍵都視為正負電荷相吸的純離子鍵,配位基 (ligand) 的孤對電子視為負的點電荷,或是部分負的電偶極;帶正電的中心金屬離子處於負電荷配位基所形成的晶體場中。當配位基加進來形成錯合物後,由於受到晶體場的交互作用,五種 d 軌域的能量會受到影響,此謂 d 軌域開裂。
錯合物的空間構型也會影響配位體形成的晶體場,像是平面四邊形、正四面體或是正八面體,中心金屬離子的 d 軌域受到的排斥力都不同,造成不同的開裂情況。
在八面體的錯合物中,d 軌域會分裂成兩個較高 (eg) 和三個較低 (t2g) 的簡併 (degenerate) 軌域,兩者之間的能量差稱為分裂能 (ligand-field splitting parameter, Δ)(圖一)。若分裂能量差恰為可見光的能量範圍,錯合物就會呈現顏色。起因於空間構型,使得d軌域中,dz2和dx2-y2這兩個軌域的電子雲集中在有配位基負電荷的位置上,斥力大,因此能量提升,提升量為0.6Δ,這兩個等能量的軌域稱為 eg 軌域,而dxy、dyz和dxz在空間中與配位基的位置剛好錯開,能量比 eg 低,比原來未配位的 d 軌域能量低了0.4 Δ,這三個等能量的軌域稱為 t2g 軌域(圖二)。
化學傳記:2001年諾貝爾化學獎得主-卡爾•巴里•夏普萊斯(K. Barry Sharpless)
國立臺灣師範大學化學系博士生三年級 林欣慧

圖一、卡爾·巴里·夏普萊斯 (來源:參考資料2)
卡爾·巴里·夏普萊斯1,1941 年 4 月 28 日生於美國賓州費城。1963 年於達特茅斯學院取得學士學位,1968 年在塔梅爾蘭教授 (Prof. Eugene E. Van Tamelen)3 的指導下於史丹福大學取得博士學位,主要研究領域為有機化學。隨後在柯爾曼教授的實驗室擔任博士後研究員 (Prof. James P. Collman)4,研究領域為有機/無機化學;1969 年轉往哈佛大學康拉德·布洛赫教授 (Prof. Konrad E. Bloch)5 實驗室進行酶學的研究。
軟硬酸鹼學說(Hard and Soft Acids and Bases)
國立臺灣師範大學化學系三年級 趙崇瀚

圖一 Ralph G. Pearson,1914年出生於美國芝加哥。(來源:參考資料4)
軟硬酸鹼學說 (Hard and Soft Acids and Bases, HSAB) 是於 1963 年,美國科學家拉爾夫 G. 皮爾森(Ralph G. Pearson,圖一)所提出一種對於路易士酸鹼的定性描述,其內容在於將路易士酸及路易士鹼分為「軟」及「硬」兩類,再依循「軟的路易士鹼偏向與軟的路易士酸反應、硬的路易士鹼偏向與硬的路易士酸反應」此一原則,來討論路易士酸鹼反應的趨向、產物的穩定性。1
霍納-沃茲沃思-埃蒙斯烯烴合成反應 (Horner-Wadsworth-Emmons Olefination)
國立臺灣師範大學化學系博士生 林欣慧
由天然物所發現的藥物分子結構中有許多的官能基,碳 – 碳雙鍵即是其中常見的一種。因此,對於如何合成碳 – 碳雙鍵就成為了科學家所注重的問題。在現今的化學界中已有許多成熟的碳-碳雙鍵合成方法,諸如:威悌反應 (Wittig reaction)、霍夫曼消去反應 (Hoffmann elimination) 等多種反應,以及今天要介紹的霍納-沃茲沃思-埃蒙斯烯烴合成反應 (Horner-Wadsworth-Emmons olefination, HWE olefination)(圖一)1,2。
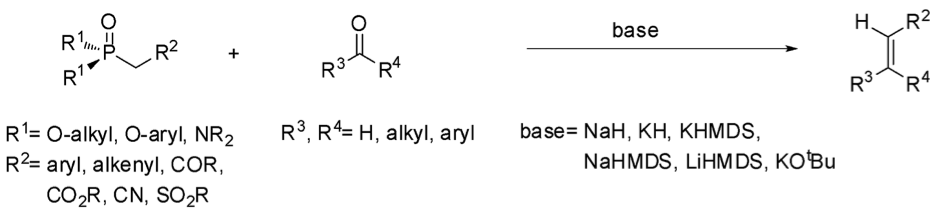
圖一 霍納-沃茲沃思-埃蒙斯烯烴合成反應(來源:作者繪製)
HWE烯烴化反應是由Leopold Horner教授於1958年首先發表,而後在1965年由William S. Wadsworth、William D. Emmons兩位教授再進行研究完善的成果。
理想氣體和凡得瓦爾氣體的比較-以二氧化碳為例
(Comparison of ideal gas and van der Waals gases – a case study in carbon dioxide)
國立臺灣師範大學化學系兼任教師 邱智宏
初學物理化學 (physical chemistry) 時,理想氣體 (perfect gas) 如影隨形,無時不在,隨時出現在各個不同的章節。由於理想氣體假設其氣體粒子不具有體積、粒子間没有吸引力、彼此間的碰撞為彈性碰撞,因此其 \(p \cdot V \cdot T\) 間的關係,可以簡潔的以 \(pV=nRT\) 加以描述。
然而真實氣體究竟佔有體積,彼此具有吸引力,碰撞時也非彈性碰撞,因此其許多特性和理想氣體不一樣,例如低溫高壓下,真實氣體大大偏離理想氣體、能被液化、有特殊的臨界點 (critical point)⋯ 等。歷來許多科學家總希望由簡潔的理想氣體方程式出發,企圖能找到一個足以說明真實氣體的方程式,其中凡得瓦爾方程式 (van der Waals equation) 就是一個很好的例子,在數學上雖然稍微複雜一些,但卻能解釋很多真實氣體的現象。
本文試著比較二種方程式的異同,並由其相異之處,解釋為何凡得瓦爾方程式更能接近真實氣體的理由。另外,以二氧化碳為例,觀察其相圖的變化情形,並說明凡得瓦爾方程式可信及不足之處。
併發聯繼催化(Concurrent Tandem Catalysis)
國立臺灣師範大學化學系碩士班一年級 薛園馨
在講究效率與原子經濟 (Atom Economy) 的現在,一鍋化 (one pot) 的合成策略可以達到減少溶劑、省下繁複的純化時間與管柱層析時所使用的沖堤液、避免因純化步驟中造成產物流失而使產率下降等等的優點,是合成化學家努力的目標,這次要介紹的併發聯繼催化 (Concurrent Tandem Catalysis) 便是其中一種方法。
併發聯繼催化指的是在單一個反應容器內有兩個或多個催化循環結合使反應物順利的進行多步反應來產生預期的產物,這當中必須考慮到每個催化劑與基質、與中間產物的選擇性才能有好的產率,併發聯繼催化可細分為以下幾種主要類型。1
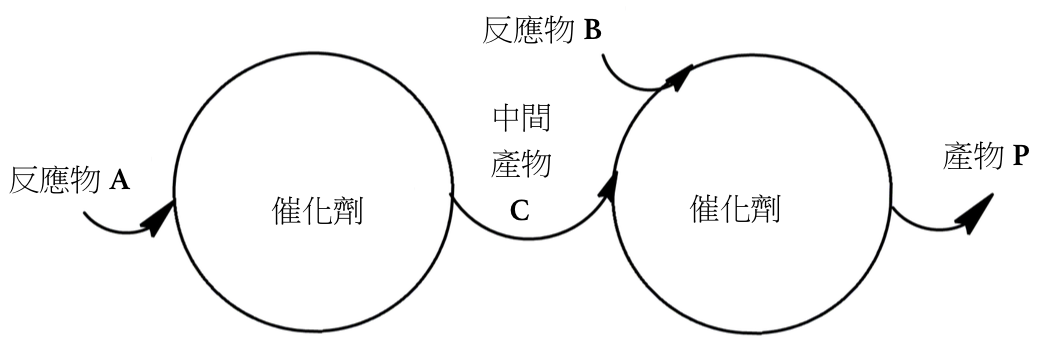
圖一、第一型的反應(作者繪製)
如圖一,反應物經催化劑甲催化後產生中間產物 C,中間產物 C 在與反應物 B 經催化劑甲催化下得到產物 P。1如圖二,首先銥金屬配位在反應物 4-戊炔-1-胺(I) 的參鍵上,配位後參鍵被氮的孤對電子攻擊而進行分子內合環反應,再脫去金屬得到的中間產物 2-甲基吡咯啉(II),此時溶劑內的三乙基矽烷 (III) 與中間產物 (II) 經過銥金屬催化下進行矽氫加成反應 (Hydrosilylation),得到最終產物含矽的吡咯烷 (IV)。2
卡爾-費雪滴定 (Karl Fischer Titration)
國立臺灣師範大學化學系 林欣慧博士三年級
卡爾-費雪滴定 (Karl Fischer Titration) 是檢測物質所含微量水分的方法,卡爾∙費雪博士於 1935 年發表於 Angewandte Chemie 期刊,利用碘、二氧化硫與水進行氧化還原反應來偵測水的含量。此滴定法可應用在多種類的固體、有機液體的水分檢測,因此目前在工業界中檢測微量水分的方法都是利用此法。反應需在非極性的溶劑中進行,不可以在酸性或鹼性溶劑中,其主要反應式為式一。1,2
I2 + SO2 + 2H2O → 2HI +H2SO4 (式一)
而碘對水的化學計量會受到溶劑中的酸鹼影響,會由 1:2 變成 1:1,因此卡爾∙費雪博士為了穩定其劑量比及促使反應的平衡向右移動,在反應系統裡加入了吡啶 (pyridine) 做為緩衝以及使用無水甲醇作為反應溶劑。大量的吡啶可與碘及二氧化硫形成鹽類再與水進行反應,得到吡啶∙碘錯合物 (Pyridium iodide, C5H5N∙HI)、亞硫酸吡啶鹽 (Pyridium sulfite, C5H5N+∙SO–3)(式二)。